Cucurbit Genetics Cooperative Report 22:43-46 (article 16) 1999
Ying Li
School of Plant, Statistical and Ecological Sciences, 111 Poole Agricultural Center, Clemson University, Clemson, SC 29634
John F. Whitesides
School of Animal & Veterinary Sciences, C123 Poole Agricultural Center, Clemson University, Clemson, SC 29634
Bill Rhodes
School of Plant, Statistical and Ecological Sciences, 111 Poole Agricultural Center, Clemson University, Clemson, SC 29634
Introduction
To analyze the DNA of plant cell populations, it is necessary to prepare a homogenous sample of nuclei. Preparation of nuclei from in vitro samples of watermelon is desirable because the choice of genotype and ploidy level at this stage saves greenhouse labor and materials. Intuitively, cells and nuclei prepared from micropropagules should be more easily extracted and prepared in pure form than from older plants with more cell wall material and more vascular tissue. In an experiment to determine optimal tetraploid generation in vitro (see Li et al, this report), we also evaluated nuclear preparation techniques.
Methods
Treated buds (Li et al, 1999) were placed on MS medium plus 3% sucrose in 0.7% agar with 10 μlM BA. The shoots were subcultured every 30-40 days.
Nuclear isolation procedure came from authors 1, 2, 3, 4, 5, and 7 with modifications. In vitro leafy shoot samples, 0.01-2.00g from the fresh clonal cultures, were excused and weighed for cell flow cytometry. The samples were immediately immersed in PBS (20 mM phosphate buffer, 154mM sodium chloride, pH 7.2=7.4) in a polypropylene tube (100×7.5mm) for 1-36 hours at 4 C. For releasing intact nuclei, different time periods of chopping or blending were compared. In the chopping technique, single edge razor blade was used to manually chop the plant material in a plastic petri dish (100 x 15 mm) with 0.5 ml PBS solution on ice.
In the blending procedure, a blender (Tissue TearorTM, Model 985-370 type 2, variable speed 5,000-30,000rpm, Biospec Products, Inc.) was used on the plant material in a plastic petri dish (100x15mm) with 0.5 ml PBS solution on ice. For releasing intact nuclei, different time periods of chopping and blending were compared. Then the mixture was filtered through 2 layers of lens paper or 50 μm nylon mesh. The nuclei and cell debris were washed with 2 ml PBS, and the filtrate was stored at 4 C for 12-24 hr or used immediately. The filtrate was centrifuged at 180xg for 1 min to pellet intact cells and cell clumps at 4C. The supernatant was poured into another clean tube (100×7.5mm) and centrifuged again at 180xg for 10 min to pellet relatively purified nuclei. The nuclei pellet was resuspended in 200 μl solution [prepared in 15 ml PBS + 15 mg dithiothreitol (DTT) + 375 μl Triton X-100 (10%w/v + 60 μl PI (5 mg/ml) + 4 μl RNaseA (DNase-free, 10 mg/ml)]. The suspension was then blended and incubated at 37 C for 15 min. All manipulations were carried out on ice or at 4 C except for the RNA digestion. To confirm whether nuclei were released and their product, each each step was examined visually under an Olympus CH-2 phase-contrast or fluorescent microscope.
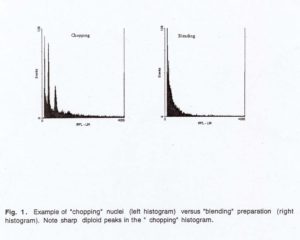
After incubation, the prepared samples were analyzed on an EPICS 751 flow cytomater (Coulter Corporation, Hialeah, FL) equipped with a data adquisition system. Excitation of PI was provided by the 488 nm line (100mW) of an arg9n laser (Model I-90, coherent) and the red fluorescence emitted by PI was collected through a 635 nm band pass filter. Chicken red blood cells, with nuclear DNA content of 2.6 pg, were used as an internal standard. Samples of 20,000 nuclei were analyzed and the data were represented as histograms (Fig. 1). Only data collected from samples with G1/G0 peaks with coefficient of variations (cvs)< 5% were used in C-value estimates (3).
The mixture was filtered through 2-layer lens paper or 50 μm nylon mesh. the nuclei and cell debris were washed with 2 ml PBS, and the filtrate was stored at 4 C for 12-24 hr or used immediately. The filtrate was centrifuged at 180g for 1 min to pellet intact cell and cell clumps at 4 C. The supernatant was poured into a clean tube (100c7.5mm) and centrifuged again at 180g for 10 min to pellet relatively purified nuclei. The nuclei pellet was resuspended in 200 μl solution (prepared in 15ml PBS + 15mg dithiothreitol + 375 μl Triton X-100(10%w/v) + 60 μl PI (5 mg/ml)). The suspension was mixed and incubated at 37C for 15 min. The prepared samples were analyzed on an EPICS 751-flow cytometer (Coulter, FL) with a Cicero acquisition module and cyclops analysis software (Cytomation, CO) with the 488 nm line (100 mw) of an argon ion laser (Model I-90, coherent). The red fluorescence emitted by PI was collected through 635 nm band pass filter. Chicken red blood cells, with nuclear DNA content of 2.6 pg, were used as an internal standard. Samples of 20,000 nuclei were analyzed and the data represented as histograms.
Results
The amount of leaf sample and the method of preparing the nuclei were found to be critical. If the quantity was too small, the quantity of nuclei were not enough to proceed with the flow cytometric analyses. If the quantity was large, the extra nuclei were wasted. Intact nuclei are released much more efficiently by chopping than blending regardless of the time period (Table 1). A period of more than 3 hours but less than 24 hours was sufficient to release the nuclei into PBS without loss of their integrity.
Table 1. Effect of nuclear isolation techniques on yield of DNA from in vitro leafy shoots.
| Method | Tissue (g) | Cell Disruption Time (min) | Yield, intact nucleiz |
| Fine Chopping | 0.01 | < 1 min | 7 |
| 0.03 | < 1 min | 18 | |
| 0.05 | 1-2 min | 55y | |
| 0.07 | 1-2 min | 67y | |
| 0.01 | 3-4 min | 72y | |
| 0.50 | 4 min | 66y | |
| 1.0 | 5 min | 70y | |
| 0.50 | 4 min | 66y | |
| 1.0 | 5 min | 70y | |
| 2.0 | >7 min | 54y | |
| Blend | 0.05 | 30 sec. speed 1 | 0 |
| 0.05 | 30 sec. speed 2 | 3 | |
| 0.05 | 30 sec. speed 3 | 7 | |
| 0.05 | 1 min, speed 1 | 4 | |
| 0.05 | 1 min, speed 2 | 3 | |
| 0.05 | 1 min, speed 3 | 15 | |
| 0.05 | 2 min, speed 1 | 20 | |
| 0.05 | 2 min, speed 2 | 17 | |
| 0.05 | 2 min speed 3 | 0 | |
| 0.05 | 3 min,speed 1 | 32 | |
| 0.05 | 3 min, speed 2 | 12 | |
| 0.05 | 3 min, speed 3 | 0 | |
| 0.05 | 4 min, speed 1 | 28 | |
| 0.05 | 4 min, speed 2 | 9 | |
| 0.05 | 4 min, speed 3 | 0 | |
| 0.05 | 5 min, speed 1 | 22 | |
| 0.05 | 5 min, speed 2 | 3 | |
| 0.05 | 5 min, speed 3 | 0 |
zThe yield was based on the average number of nuclei in 5 different ocular fields under a phase contrast microscope (5 x 40).
y Sharp peak appeared during flow cytometry.
Acknowledgements. The flow cytometer was made available through the College of Agriculture, Forestry and Life Sciences, and the South Carolina Experiment Station.
Literature Cited
- Arunmuganathan, K. and e. Earle. 1991a. Estimation of nuclear DNA content of plants by flow cytometry. Plant Mol. Biol. Reporter 9:229-241.
- Arunmuganathan, K. and E. Earle. 1991b. Nuclear DNA content of some important plant species. Plant Mo. Biol. Reporter 9:208-218.
- Baird, W.V., A.S. Estager and J.K. Wells. 1994. Estimating nuclear DNA content in peach and related diploid species using laser flow cytometry and DNA hybridization. J. Amer. Soc. Hort. Sci. 11:1321-1316.
- Michaelson, M., J. Price, J. Elison and Johnston. 1991. Comparison of plant DNA contents determined by Fuelgen microspectrophotometry and laser flow cytometry. Amer. J. Bot. 78:183-188.
- Mysore, K.S. and V. Baird. 1997. Nuclear DNA content in species of Eleusine (Gramineae): a critical re-evaluation using laser flow cytometry. Pl. Syst. Evol. 207:1-11.
- Tosca, A., R. Pandolfi, S. Citterio, A. Fasoli, and S. Sgorbati. 1995. determination by flow cytometry of the chromosome doubling capacity of colchicine and oryzalin in gynogenetic haploids of Gerbera. Plant Cell Reports 14:455-458.
- Zhang, X.P., B.B. Rhodes, H.T. Skorupska and W. C. Bridges. 1994a. Determination of watermelon ploidy level using flow cytometry. Cucurbit Genetics Coop. Rpt. 17:102-105.
- Zhang, X.P., B.B. Rhodes and J.W. Adelberg. 1994b. Shoot regeneration from immature cotyledons of watermelon. Cucurbit Genetics Coop. Rpt. 17:111-115.