Cucurbit Genetics Cooperative Report 14:25-28 (article 10) 1991
L. D. Knerr and J. E. Staub
Vegetable Crops Research, USDA/ARS, Department of Horticulture, University of Wisconsin, Madison 53706
Genetic diversity in the U.S. Cucumis sativus var. sativus L. and var. hardwickii (R.) Alef. germplasm collection was assessed using 18 putative enzyme coding loci (2). These loci include Gpi-1, Gpi-2, Gr-2, G2dh, Idh, Mdh-1, Mdh-2, Mdh-3, Pepla-2, Peppap-2, Per-4, Pgd-1, Pgd-2, Pgm-1, Pgm-3, and Skidh-2. Three types of multivariate analyses were utilized to depict affinities and similarities among individual plant introductions (PIs) and PIs grouped by geographic region. Cluster analysis (compact linkage method) was used to group PIs by geographic region. PIs with similar isozyme phenotypes were placed in close proximity on the resulting dendrogram (Fig. 1). Principal component analysis (PCA) discriminated among individual PIs. A third procedure, classification and regression tree (CART) analysis identified enzyme loci (Gr-1, Mpi-2, Pepla-2, Pgd-2, Pgm-1, and Skidh-2) which were most discriminating in the analysis.
Inheritance and linkage studies (1) revealed that variation at Gpi-2, Gr-1, Pgm-3 and Skdh-2 did not have predictable genetic bases and therefore this variation could not be classified as allozymic. This dictated that a multivariate re-evaluation omitting these 4 loci be conducted to more accurately describe the allozymic variation present in the U.S. germplasm collection.
The removal of data affected the classification of PIs which possessed putative rare alleles. The impact of rare alleles is important on the analysis and the removal of the putative rare (Gpi-2, Gr-2, Pgm-3, and Skdh-2) variants affected the results of principal component and cluster analysis partially by placing a greater weight on each of the remaining 14 loci in the re-evaluation.
PCA reduced the revised data set and identified 176 (a 77% reduction) different enzyme phenotypes as opposed to the 238 (a 68% reduction) in the initial analysis (2). This reflects a considerable decrease in the original amount of variability. PCA also grouped PIs according to their overall variability. Table 1 contains a partial listing of PIs according to the first principal component. PIs listed farther apart from one another. A complete listing of the entire U.S. C. sativus germplasm collection according to overall variability as determined by the first principal component, as well as the allozyme phenotypes for all PIs evaluated, can be found in the U.S. Germplasm Resources Information Network.
The CART analysis results remained unchanged with the exception of the data elimination. Mpi-2, Pepla-2, Pgd-2 and Pgm-1 are the mos discriminating loci. The ramifications of this result are that initial screening of additional germplasm might only include these loci in order to save time and expense.
Reanalysis of the data using cluster analysis resulted in changes in geographical relationships (Figure 2). Examples and explanations for some of these changes are:
1) In the original dendrogram Indonesia and Iraqi accessions were depicted to be more distinct from the rest of the collection. In the new dendrogram these distinctions disappear. Both of these countries possessed accessions with putative alleles which were eliminated after re-evaluation. Similar conformational changes occurred with Thai and Australian accessions.
2) Since hardwickii possessed alleles for Per-4 and Idh which were not present in the remainder of the collection, an initial node separates it from the remaining sativus accessions. The removal of non-variable loci apparently put a greater weight on the remaining loci, allowing a more accurate description of relations. A similar event occurred with Polish accession in which G2dh variation was unique. Certain countries, such as Egypt and Hungary, were partitioned into unique nodes due to the occurrence of multiple alleles that existed a low frequencies.
Due to unknown ancestry of the collection and the sharing of germplasm among countries, geographic affinities described by clustering procedures require judicious appraisal. As this data set expands with the addition of polymorphic allozyme coding loci and their linkage relations are estimated, a more accurate depiction of relationships will be possible.
Figure 1. Cluster analysis (compact linkage method) of Cucumis sativus L. plant introductions grouped by country using 18 enzymes loci as framing criteria.
MAXIMUM LINKAGE DISTANCE
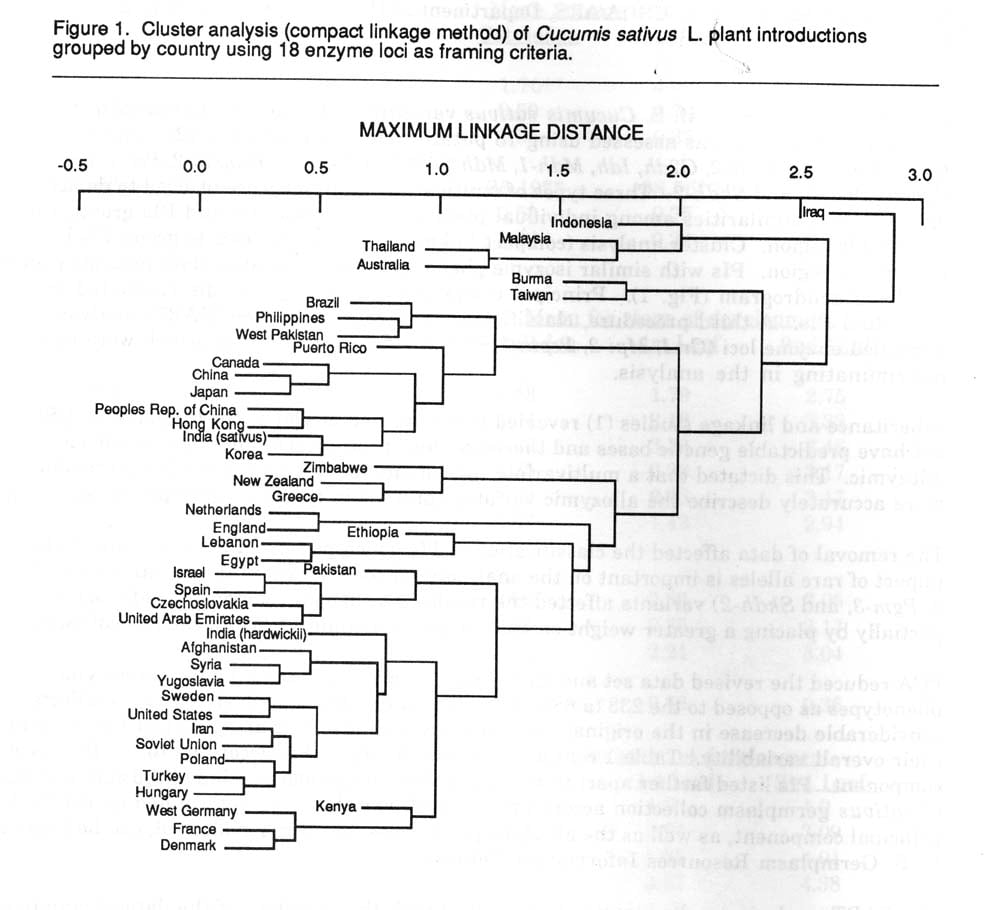
Figure 2. Cluster analysis (compact linkage method) of Cucumis sativus L. plant introductions grouped by country using 14 allozyme coding loci as framing criteria.
MAXIMUM LINKAGE DISTANCE
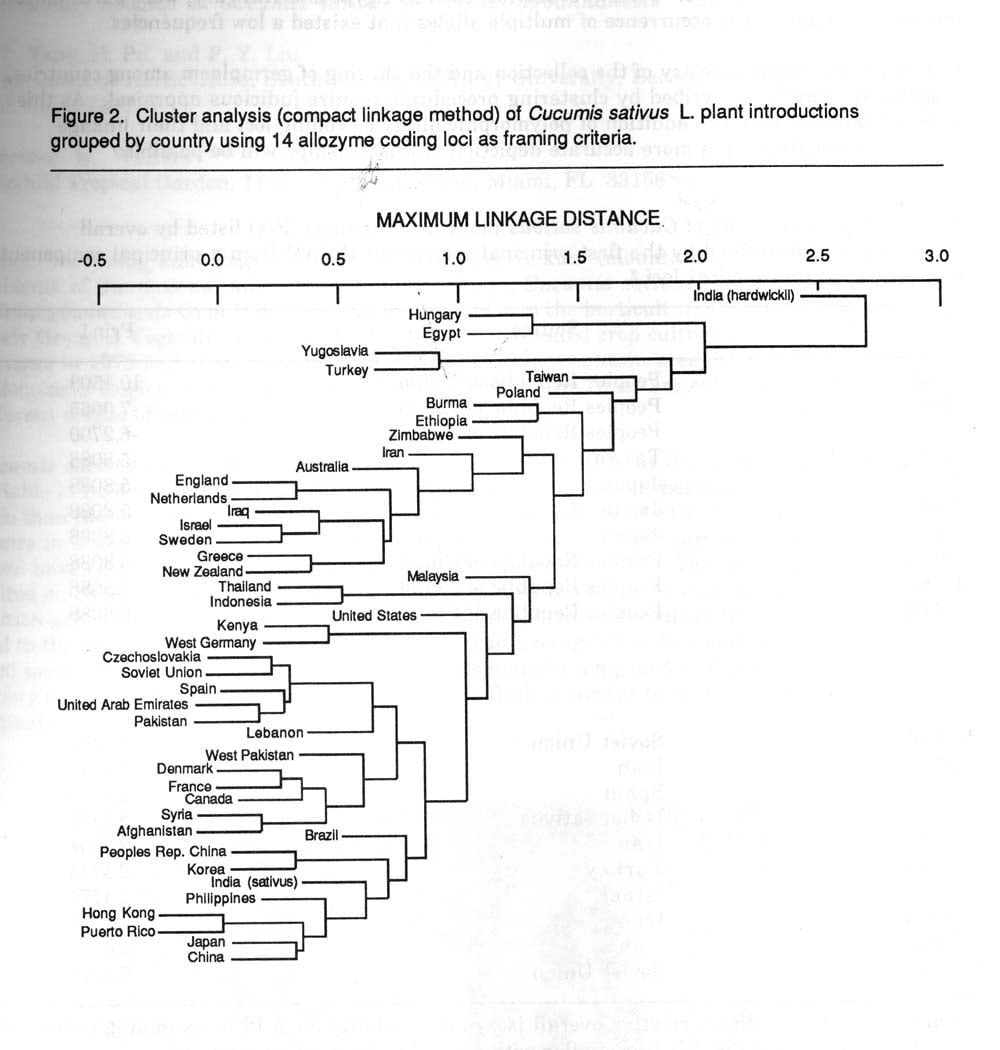
Table 1. A partial listing of Cucumis sativus plant introductions (PIs) listed by overall variability as determined by the first principal component (Prin1) from a principal component analysis of 14 biochemical loci.z
| PI accession | Source | Prin 1 |
| 432851 | People’s Republic of China | -10.1509 |
| 432854 | People’s Republic of China | -7.0065 |
| 432858 | People’s Republic of China | -6.2700 |
| 321007 | Taiwan | -5.8088 |
| 390243 | Japan | -5.8088 |
| 390246 | Japan | -5.8088 |
| 390257 | Japan | -5.8088 |
| 430585 | People’s Republic of China | -5.8088 |
| 432850 | People’s Republic of China | -5.8088 |
| 432853 | People’s Republic of China | -5.8088 |
| 458855 | Soviet Union | 2.2471 |
| 227013 | Iran | 2.2914 |
| 257286 | Spain | 2.3130 |
| 164734 | India, sativus | 2.3177 |
| 211984 | Iran | 2.3904 |
| 169353 | Turkey | 2.4774 |
| 292012 | Israel | 2.4774 |
| 344434 | Iran | 2.4774 |
| 344444 | Iran | 2.4774 |
| 458856 | Soviet Union | 2.4774 |
z Values for Prin1 indicate relative overall isozyme variability for a PI in ascending order. PIs with similar values for Prin1 are similar with respect to isozyme phenotypes.
Literature Cited
- Knerr, L. D. and J. E. Staub. 1992. Inheritance and linkage relationships of allozyme-coding loci in cucumber (Cucumis sativus L.). Theor. Appl. Genet. (submitted).
- Knerr, L. D., J. E. Staub, J. D. Holder and B. P. May. 1989. Genetic diversity in Cucumis sativus L. assessed by variation at 18 allozyme coding loci. Theor. Appl. Genet. 78:119-128.