Cucurbit Genetics Cooperative Report 15:35-39 (article 13) 1992
S.J.T. Raharjo and Z.K. Punja
Department of Biological Sciences, Simon Fraser University, Burnaby, British Columbia, Canada V5A 1S6
Methods for the culture and sustained cell divisions and growth of plant cells or cell aggregates in liquid medium containing appropriate growth regulators have been reported for a number of important crop species, including carrot (1), bell pepper (12), white poplar (9), tepiary bean (6) and cotton (4). In cucumber (Cucumis sativus L.), several methods for initiating suspension cultures and regenerating plantlets from several fresh market cultivars have been described (2, 3, 7). The frequency of regeneration from those suspension cultures was affected by genotype and the growth regulators used (2, 7), necessitating the modification of procedures for different cultivars.
In general, suspension cultures offer several advantages over plant tissue growth on solid media. Since the cells and developing embryos are evenly exposed to nutrients and growth regulators in the liquid medium, the effects of any gradients can be eliminated, allowing more synchronous control over plantlet development. Second, the cell clusters and embryos usually separate from each other in the medium, allowing easy handling and recovery. Third, the number of potential plantlets which can be obtained from a single culture can be large if the conditions are optimized, thereby achieving exponential increases in plant numbers. Fourth, by exposure to specific selection pressure (for example toxins and other chemicals), cell lines could be recovered which display an enhanced level of tolerance.
Plant material. Seeds (provided by Campbell Inst. Res. & Tech.) of the pickling cucumber cultivar Endeavor, a gynoecious hybrid (WI 2870G X Clinton) were soaked in water for about 30 min, and the seed coat was removed carefully using a sharp scalpel. This was followed by surface-sterilization in 70% ethanol for 30 sec. a 4 min soak in 10% solution of commercial bleach (Javex, 6.25% sodium hypochlorite) and three rinses in sterile distilled water. Seedlings were grown in Magenta boxes (Magenta Corp., Chicago, IL) containing 50 ml of half-strength Murashige and Skoog (MS) basal medium (8), with 100 mg 1-1 ampicillin to minimize bacterial contamination. To ensure uniform germination, the boxes were incubated in the dark at a temperature of 29 ˚ C with a 16 h*day-1 photoperiod.
Initiation of callus. Petiole segments 3 to 5 mm long from seedlings grown in vitro were used as the explant source. The medium used for callus initiation was full-strength MS (8) (with full complement of major and minor salts, vitamins, 30 g*1-1 sucrose), with 100 mg*1-1 ampicillin. The medium was supplemented with 2,4-D/BA at 5/5 μ M and solidified by addition of 10 mg *1-1 Sigma tissue culture agar. The pH was adjusted to 5.8 prior to autoclaving at 15 psi for 20 min. Approximately 25 ml of medium was dispensed into disposable Petri dishes (20 X 100 mm). Eight explants were placed in each Petri dish, and the dishes were sealed with Parafilm (R). All dishes were incubated in the dark for 3 weeks prior to placing in the light at an intensity of 300 μ Em-2s-1 provided by cool-white fluorescent lamps), 16 h*day-1 photoperiod, and at ambient temperatures of 25 to 28 ˚ C.
Initiation. Eight weeks after explants were plated, creamy-yellow embryogenic calli which developed were dissected and used as the donor of cells or cell aggregates for suspension culture utilizing liquid MS medium containing full strength major and minor salts, vitamins, and 30 g*l-1 sucrose. Several combinations of growth regulators were evaluated NAA/BA (1.0/1.0 μ M), NAA/Z (1.0/1.0 μ M), NAA/adenine-SO4(1.0/200 μ M), 2,4-D/BA (1.0/1.0 μ M), and 2,4-D (1.0 μ M). Embryogenic callus from one explant was added to each 250 ml Erlenmeyer flask containing 75 ml of medium. There were three flasks for each combination of growth regulator tested. The flasks were kept in dim light at ambient temperatures of 24 to 28 ˚ C and shaken continuously at 120 rpm (on a gyratory shaker). After 4 weeks, the condition of the suspension cultures was noted and healthy-appearing cultures were subsequently subcultured into fresh medium containing the suspension culture through Sigma cell sieve (with 0.38 mm openings), followed by two rinses of the cells/aggregates using liquid MS medium without growth regulators and transferring the cells/aggregates into the fresh medium.
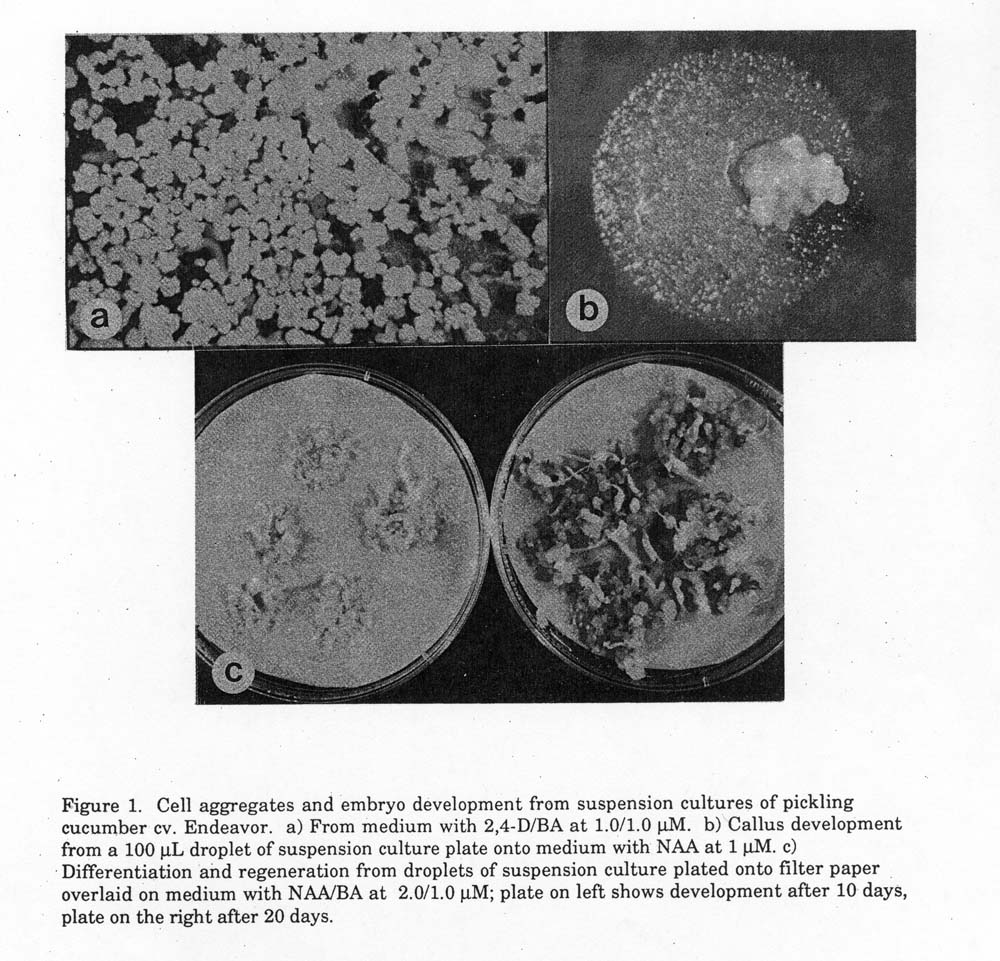
Maintenance. Two weeks after the first cubculture, suspension cultures initiated in medium with 2,4-D/BA (1.0/1.0 μ M) showed the best appearance. The best cultures produced a large number of yellow cells/aggregates, did not result in formation of roots or embryo elongation, and did not show evidence of browning (Fig. 1a). Only those suspension cultures were maintained further. The suspension cultures were subcultured every 2 to 3 weeks as follows. The flasks were removed from the shaker, the cells/aggregates were allowed to settle down to the bottom of the flask; then about 10 ml of the suspension (with dense cells/aggregates) was pipetted (using a volumetric pipet of 10 ml capacity with a large-holed tip) into 75 ml of fresh medium with 2,4-D/BA (1.0/1.0 μ M) and the flask was shaken manually. After all of the cells/aggregates had settled, 10 ml was once again pipetted and dispensed into each of 4 to 5 flasks with 75 ml of fresh maintenance medium containing 2,4-D/BA (1.0/1.0 μ M). The suspension cultures were maintained on a gyratory shaker at 120 rpm, at the same conditions as those for their initiation.
Regeneration. The first attempt to induce regeneration was carried out using 5-month old cultures by planting 1 ml of the suspension culture, two weeks after last subculture, onto MS medium without any growth regulators (MS0) or MS0 with activated charcoal (0.5%), or five 100 μ l drops were plated on MS with NAA (1 μ M) (Fig. 1b). However, this procedure did not induce formation of normal plantlets. A second procedure similar to that described by Bergevoet et al. (2) was tested with 11-month old cultures. All the cells/aggregates from each flask 10 days after the last subculture, were pipetted and transferred into another flask containing 100 ml liquid MS medium without growth regulator and shaken manually. After all of the cells/aggregates had settled, approximately 10 ml of the medium with dense aggregates was pipetted out. To plate the aggregates, five 100 μ l drops of the suspension were plated onto filter paper (Whatman) overlaid onto solid regeneration medium containing either IAA/kinetin ( 0.1/1.0 μ M) or NAA/BA (2.0/1.0 μ M) (Fig. 1c).
Results. Petiole segments initiated callus after about one week on medium containing 2.4-D/BA (5/5μ M) and the callus developed embryogenic (yellow and friable) sectors and embryos 5 to 7 weeks later. This embryogenic callus was dissected and transferred into liquid medium to initiate the suspension culture. After 2 to 3 weeks of shaking on a gyratory shaker, callus in the medium with 2,4-D/BA (1.0/1.0 μ M) started to break apart, forming suspension of cells/aggregates (Fig. 1a). In the medium containing either of NAA/BA (1.0/1.0 μ M), NAA/Z (1.0/1.0 μ M), NAA/adenine-SO4 (1.0/200 μ M), or 2,4-D (1.0 μ M), the callus did not break apart or formed elongated and rooted embryos, resulting in browning of the tissue or the liquid after 4 weeks. The medium containing 2,4D/BA (1.0/1.0 μ M also appeared to be most appropriate for long-term maintenance of the suspension culture. Using this medium, healthy and regenerable suspension cultures have been maintained for more than 15 months. A subculture every 2 to 3 weeks appeared to be optimal for long-term maintenance of the suspension culture; after more than 3 weeks, the aggregates increased in size and developed elongated and rooted embryos, resulted in the browning of the liquid and rapid decline of the culture. Aggregates from this suspension culture were able to form normal shoots when plated onto filter paper overlaid on medium containing NAA/BA (2.0/1.0 μ M) (Fig. 1c). Shoots developing on regeneration medium were excised after 3 weeks and transferred onto MS medium without growth regulators where the shoots elongated and developed roots. Although the first leaves appeared distorted, subsequent leaves usually looked normal. The frequency of plantlet formation is given in Table 1. About 20 plantlets were obtained from each Petri dish containing 500 μl of the initial suspension.
Discussion. Procedures to regenerate plantelets from suspension cultures of different genotypes of cucumber have included various techniques, such as direct-plating of embryoid aggregates onto MS medium (solid, semi-solid, liquid) without growth regulators (7), plating the aggregates on MS medium containing activated charcoal or germinating the embryos from suspension culture in MS basal medium (after a pre-wash in MS medium containing activated charcoal (3), and plating the aggregates on MS medium with IAA and kinetin (2). The addition of activated charcoal, which was useful for initiating regeneration from callus as well as suspension cultures in other studies (3,5), was not effective in this study. That might be due to the fact that our suspension culture had ben maintained over a longer period due to genotype differences.
In our study, MS medium with IAA/kinetin (0.1/1.0 μ M) gave regeneration of adventitious shoots through organogenesis. However, the shoots dedifferentiated very quickly prior to the stage where they could be transferred to MS free of growth regulators (for plantlet development). The use of NAA/BA at 2.0/1.0 μ M in the plating medium provided the highest recovery of plantlets in this study. That combination of growth regulators has also been used in the regeneration of a range of pickling cucumber cultivars from explants (11) and from protoplasts (10). The procedure described in this study may be applicable to regeneration from suspension cultures of other cultivars of pickling cucumber. In preliminary studies, the cultivar Calypso has also responded in a similar manner.
Table 1. Regeneration and appearance of regenerants of Cucumis sativus cv. Endeavor 3 weeks after plating aggregates from suspension culture onto regeneration medium.
No. plantlets/dish |
|||
Plating medium |
Total |
Normal |
Appearance |
| MS0z | 0 | 0 | aggregates forming calli, embryo dedifferentiation, rooting, browning |
| MS+charcoal (0.5%)z | 0 | 0 | aggregates forming calli, embryo dedifferentiation, rooting, browning |
| MS + NAA (1 μ M)y | 18 | 0 | aggregates forming calli and shoots, shoot dedifferentiation |
| MS + NAA/BA (2.0/1.0 μ M)x | 38 | 21 | aggregates forming calli and normal shoots from embryos, less shoot dedifferentiation, rooting |
| MS + IAA/kinetin (0.1/1.0 μ M)x | 35 | 0 | aggregates forming calli and shoots, shoot dedifferentiation, rooting |
z One ml of suspension cultures was plated onto each dish of medium, 14 days after the last subculture.
y Five 100 μ l drops of suspension culture were plated onto each dish of medium, 10 days after the last subculture.
x Five 100 μ l drops of dense cell/aggregate suspension were plated onto filter paper overlaid onto medium, 10 days after the last subculture.
Figure 1. Cell aggregates and embryo development from suspension cultures of pickling
cucumber cv. Endeavor. a) From medium with 2,4-D/BA at 1.0/1.0 µM. b) Callus development from a 100 µL droplet of suspension culture plate onto medium with NAA at 1 µM. c) Differentiation and regeneration from droplets of suspension culture plated onto filter paper overlaid on medium with NAA/BA at 2.0/ 1.0 µM; plate on left shows development after 10 days, plate on the right after 20 days.
Literature Cited
- Ammirato, P.V. Induction, maintenance, and manipulation of development of embryogenic cell suspension cultures. In: Cell Culture and Somatic Cell Genetics of Plants, Vol. 1. (I.K. Vasil, ed.) –. 139-151. Academic Press, New York.
- Bergervoet, J.H.W., F. van der Mark and J.B.M. Custers. 1989. Organogeneis versus embryogenesis from long-term suspension cultures of cucumber (Cucumis sativus L.). Plant Cell Rept. 8: 116-119.
- Chee, P.P. and D.M. Tricoli. 1988. Somatic embryogenesis and plant regeneration from cell suspension cultures of Cucumis sativus L. Plant Cell Rept. 7: 274-277.
- Finer, J.J. 1988. Plant regeneration from somatic embryogenic suspension cultures of cotton (Gossypium hirsutum L.). Plant Cell Rpt. 7: 399-402.
- Gadasi, G. and M. Ziv. 1986. enhanced embryogenesis and plant regeneration from cucumber (Cucumis sativus L.) callus by activated charcoal in solid/liquid double-layer cultures. Plant Sci. 47: 115-122.
- Kumar, A.S., O.L. Gamborg and M.W. Nabors. 1988. Regeneration from long-term suspension cultures of tepary bean (Phaseolus acutifolius). Plant cell Rpt. 7: 322-325.
- Malepszy, S. and E. Solarek. 1986. In vitro culture of Cucumis sativus L. IV. Conditions for cell suspension. Genetica Polonica. 27: 249-253.
- Murashige, T. and F. Skoog. 1962. A revised medium for rapid growth and bioassays with tobacco tissue cultures. Physiol. Plant. 15: 73-497.
- Park, Y.G. and S.H. Son. 1988. regeneration of plantlets from cell suspension culture derived callus of white poplar (Populus alba L.). Plant Cell Rpt. 7: 567-570.
- Punja, Z.K., F.A. Tang and G.G. Sarmento. 1990. Isolation, culture and plantlet regeneration from cotyledon and mesophyll protoplasts of two pickling cucumber (Cucumis sativus L.) genotypes. Plant Cell Rpt. 9: 621-64.
- Punja, Z.K., N. Abbas, G.G. Sarmento and F.A. Tang. 1990. Regeneration of Cucumis sativus var. sativus and C. sativus var. hardwickii, C. melo, and C. metuliferus from explants through somatic embryogenesis. Plant Cell Tissue Organ Culture 21: 93-102.
- Williams, P.D., A.K. Wilkinson, J.A. Lewis, G.M. Black and F. Mavituna. 1988. A method for the rapid production of fine plant cell suspension culture. Plant Cell Rpt. 7: 459-462.